


Abstract
Loss of BRCA1 tumor suppressor function is a critical event in breast tumorigenesis. We have previously identified the stress hormone hydrocortisone as a negative regulator of BRCA1 expression in nonmalignant mammary cells. Here, we have identified a direct role for the unliganded glucocorticoid receptor (GR) in BRCA1 upregulation in the absence of hydrocortisone. The positive regulatory effect of GR is lost upon the addition of hydrocortisone. We have shown that GR interacts with the BRCA1 promoter only in the absence of hydrocortisone, and that this interaction is mediated through the β-subunit of the ets transcription factor GA-binding protein (GABP) at the RIBS promoter element. GR and GABPβ interact in both coimmunoprecipitation and mammalian two-hybrid assays, and this interaction involves the N-terminal to central regions of both proteins. This work presents the first evidence of a ligand-independent role for GR as a positive regulator of gene expression, and loss of GR from the BRCA1 promoter in response to stress hormones leads to decreased BRCA1 expression. Because low levels of BRCA1 have been implicated in the development of sporadic breast cancer, this may represent a novel mechanism through which prolonged stress signaling increases breast cancer risk. Mol Cancer Res; 10(4); 558–69. ©2012 AACR.
Introduction
Germ line mutations in the BRCA1 tumor suppressor contribute to familial breast tumor formation but BRCA1 mutations do not seem to occur in sporadic breast cancer tumors (1). Instead, sporadic breast tumors exhibit decreased levels of BRCA1 expression, and the degree of downregulation seems to be correlated with tumor grade, rate of tumor progression, and risk of metastasis (2–6). This implies that loss of BRCA1 function, either through decreased activity or downregulation of expression, is a critical event in the etiology of breast cancer. Phenotypically normal breast epithelial cells from individuals harboring a BRCA1 germ line mutation (a so called “one-hit” mutation accompanied by a 50% decrease in BRCA1 function) express an altered mRNA profile compared with normal cells from individuals without a BRCA1 mutation (7). This suggests that decreases in functional BRCA1 levels on the order of 50% are biologically significant. The importance of BRCA1 in breast cancer is attributable to its involvement in a number of regulatory processes important for maintenance of genomic stability and prevention of cell transformation (8). More recently, BRCA1 has been shown to be required for mammary stem/progenitor cell differentiation, whereby loss of BRCA1 results in expansion of the stem/progenitor population (9). Consistent with this, some genes enriched in mutant BRCA1 one-hit cells are expressed in stem and progenitor cells (7, 9). Because BRCA1 also participates in maintaining genomic integrity, loss of BRCA1 function could lead to cancer development through the production of genetically unstable stem/progenitor cells (9).
The transcriptional regulation of BRCA1 is modulated by a variety of hormones, developmental cues, and other effectors (10). We have shown that the RIBS (EcoRI Band Shift) element is required for maximal BRCA1 promoter activity and contains 2 binding sites for the ets transcription factor GA-binding protein (GABP; ref. 11). Human GABP exists as a heterodimer consisting of an ets helix-loop-helix DNA-binding domain (DBD) subunit (GABPα) and a Notch-ankyrin repeat subunit (GABPβ) which contains the activation domain as well as a domain required for the formation of tetrameric complexes (12). GABP has been implicated in the regulation of genes in response to cell growth, activation of respiration-related genes (13), as a downstream mediator of ErbB3 and ErbB4 signaling (14), and recently in connecting mitochondrial metabolism and breast differentiation (15). The interaction of the α- and β-subunits with each other and with other transcription factors defines the ability of GABP to regulate the expression of its target genes.
BRCA1 can be considered as a central rheostat of breast cancer risk, with its transcriptional regulation being controlled by a variety of factors, one of which is psychological stress. Various epidemiologic studies have indicated that psychological stress produces a significant increase in breast cancer risk, and that the nature and magnitude of the effect of stress on risk is comparable with that of other well-known risk factors for breast cancer (16). Furthermore, specific studies have suggested that the cancer-causing effects of stress may be fairly specific to the breast (17, 18). We previously presented the first molecular evidence for a connection between stress and breast cancer with our observation that BRCA1 expression is repressed by the synthetic stress hormone hydrocortisone in nonmalignant mouse mammary cells (19). This work was the first breast-specific molecular mechanism linking stress and breast cancer development.
In response to a stress signal, the primary human stress hormone cortisol binds to its intracellular receptor, the glucocorticoid receptor (GR), which is a member of the steroid receptor superfamily of nuclear receptors (20). While it has traditionally been accepted that nuclear receptors are activated in response to ligand binding, several reports exist of progesterone and estrogen receptor activation and nuclear activity even in the absence of hormone (21). GR has been reported to be activated by various stimuli in the absence of glucocorticoid ligands (21–24), and unliganded GR has recently been found to bind directly to and exert a repressive effect on the IL-6 promoter in response to TNF-α in endocervical cells (25). In addition, it has been suggested that breast cancer progression may be associated with an accumulation of GR in the cytoplasm of tumor cells (26).
In this report, we present, for the first time, evidence for a ligand-independent role for GR in the activation of BRCA1 expression in breast cells. We show that this effect is mediated through an interaction between GR and GABPβ at the RIBS element of the BRCA1 promoter, which occurs only in the absence of hydrocortisone ligand. This interaction and the associated positive regulatory effect are lost upon addition of hydrocortisone, which may represent a novel mechanism through which stress signaling increases breast cancer risk. The unliganded GR plays a previously unknown role in gene activation with profound implications for our understanding of GR-mediated processes.
Materials and Methods
Cell culture and treatments
The nontumorigenic murine mammary epithelial cell line EPH-4, and the karyotypically normal, human telomerase reverse transcriptase–immortalized mammary epithelial cell line 184-hTERT were gifts of Dr. Calvin Roskelley (University of British Columbia, Vancouver, Canada). The human breast carcinoma cell lines MCF-10A and MCF-7 were obtained from American Type Culture Collection. All cells were cultured as previously described (15, 19). Cell treatments were completed using media lacking FBS or horse serum and containing either 1 μg/mL hydrocortisone (Sigma), 1, 5, or 10 μmol/L RU-486 (Sigma), or ethanol vehicle for 24 or 48 hours.
DNA constructs
Creation of the L6-pRL, L6ΔR-pRL, L6mUPmE2F-pRL, L6ΔRmUPmE2F-pRL, RIBSn-pRL, and GF-TATA-pRL BRCA1 promoter constructs has been described previously (27).
The rat constructs wild-type GR (GRwt) and GRL501P (GR with a leucine to proline mutation at amino acid position 501, which abolishes its DNA-binding ability) were gifts of Keith Yamamoto (University of California, San Francisco, CA), and their construction has been described previously (28, 29).
Construction of the H1-2 vector has been described previously (27). To construct the shGR vector, the oligos GRshRNA5′ and GRshRNA3′ were annealed (Supplementary Table S1). The product was cut with BamHI and HindIII [New England Biolabs (NEB)] and ligated into the H1-2 vector.
The constructs PRR, PRR-M1, PRR-M2, and PRR-M3 were gifts of Dr. Calvin Roskelley.
To construct pFLAG-GRα, the GRα gene was PCR amplified from MCF-7 cDNA in 2 separate reactions. The 5′ end was amplified using primers GRα5′(+) and GRα5′(−; Supplementary Table S2). The 3′ end was amplified using primers GRα3′(+) and GRα3′(−; Supplementary Table S2). The 5′ section was cut with NotI and PstI (NEB), and the 3′ section was cut with PstI and BamHI (NEB). The 5′ and 3′ sections were simultaneously ligated together and into the p3X-FLAG (pFLAG) expression vector (Sigma).
Mammalian 2 hybrid vectors pACT and pBIND, as well as firefly luciferase reporter pG5-luc were obtained from Promega. Full-length GRα, as well as its individual domains, were PCR amplified from pFLAG-GRα using primers listed in Supplementary Table S3. Each PCR product was cut with NdeI and BamHI and ligated into pACT. Similarly, full-length GABPβ, as well as its individual domains, were PCR amplified from pFLAG-GABPβ (construction described previously; ref. 15) using primers listed in Supplementary Table S4. PCR products were cut with BamHI and NotI and ligated into pBIND.
Transient transfections and luciferase assays
Transfections were carried out as described previously (15, 19). For standard transfection assays, control cytomegalovirus (CMV)-luc vector (Promega) was used at 25 ng per well, as were GR expression vectors and empty vector controls. The remainder of the 250 ng per well was allotted to the appropriate Renilla luciferase reporter vector. For knockdown transfections, 50 ng of the short hairpin RNA (shRNA) construct or its empty vector were used. For mammalian 2-hybrid transfections, 50 ng of each pBIND-GABPβ construct was used along with 25 ng of each pACT-GR construct. For all cell lines, cells were treated with hydrocortisone, RU-486, or ethanol vehicle (as described earlier) in serum-free medium 24 hours following transfection. Either 24 or 48 hours after treatment, cells were harvested for the Dual Luciferase Assay as previously described (15).
Quantitative real-time PCR
RNA and RT products were prepared as described previously (15, 19). Quantitative real-time PCR (qRT-PCR) reactions for human BRCA1 were carried out as described previously (15), with the exception that hypoxanthine phosphoribosyltransferase 1 (HPRT1) was used as an internal control instead of TBP (primers listed in Supplementary Table S5). BRCA1 expression for each cell line was calculated relative to the results for the untreated sample for each cell line with the Pfaffl method (30). qRT-PCR reactions for mouse BRCA1 (with TBP as an internal control) were carried out as described previously (ref. 19; primers listed in Supplementary Table S6). BRCA1 expression for each cell line was calculated relative to the results for the untreated sample with the ΔΔ Ct method presented by PE Applied Biosystems (Perkin Elmer).
Immunoprecipitation assay
MCF-7 cells were transfected as described earlier with different combinations of GABPα, GABPβ, and FLAG-GRα. Cells were treated 24 hours after transfection with either ethanol vehicle or 1 μg/mL hydrocortisone in serum-free medium. Following a 24-hour incubation, whole-cell lysates were prepared in modified RIPA buffer as described previously (27). Fifty micrograms of protein was incubated with 1 μg of either anti-GABPβ (sc-28684; Santa Cruz Biotechnology Inc.) or anti-FLAG (sc-807; Santa Cruz Biotechnology Inc.) for 4 hours at 4°C. The protein–antibody mixture was incubated overnight with 20 μL of Protein A/G PLUS-Agarose Immunoprecipitation Reagent (sc-2003; Santa Cruz Biotechnology Inc.) at 4°C. Beads were washed with PBS and incubated with 50 μL of 1× SDS loading buffer at 98°C for 15 minutes to elute bound protein.
Western blotting
Lysates were prepared in 1× SDS loading buffer and analyzed by standard Western blotting procedures. Polyvinylidene fluoride membranes (Millipore) were probed with the appropriate primary antibody: anti-GR (1:500; ab3579; Abcam), anti-TBP (1:2,000; ab818; Abcam), anti-GABPβ (1:5,000; sc-28684; Santa Cruz Biotechnology Inc.), or anti-FLAG (1:1,500; sc-807; Santa Cruz Biotechnology Inc.). The secondary antibodies used included goat anti-rabbit (1:10,000; sc-2004; Santa Cruz Biotechnology Inc.) to detect GR, goat anti-mouse (1:10,000; 115-035-003; Jackson ImmunoResearch) to detect TBP, and Clean-Blot IP Detection Reagent (1:700; Thermo Scientific/Fisher) for all immunoprecipitated lysates. Secondary antibody detection was carried out by chemiluminescence (Thermo Scientific/Fisher).
ChIP assay
EPH-4 cells were plated and treated as described earlier. Chromatin immunoprecipitation (ChIP) assays were conducted with the ChIP-IT Express Enzymatic Kit (Active Motif). Each reaction was carried out using chromatin from 2 × 106 cells and 2 μg per reaction of affinity-purified antibody (or water as a negative control). The following antibodies were used: anti-GR (ab3579; Abcam), anti-GABPα (sc-22810; Santa Cruz Biotechnology Inc.), anti-GABPβ (sc-28684; Santa Cruz Biotechnology Inc.), anti-hemagglutinin (sc-805; Santa Cruz Biotechnology Inc.), anti-acetylated histone H3 (06-599; Upstate Biotechnology), anti-GST (sc-459; Santa Cruz Biotechnology Inc.), anti-Fra-2 (sc-604; Santa Cruz Biotechnology Inc.), anti-USF-2 (sc-861; Santa Cruz Biotechnology Inc.), and anti-RYBP (ab5976; Abcam). The PCR primers amplified the mouse BRCA1 promoter from position −335 to +73 [(+) 5′-TCCGGGAGCGATTCCCACCC and (−) 5′-AAGATCCGTACTTCCAAGCG]. A water blank (no template) and EPH-4 input chromatin were also subjected to PCR amplification as controls. ChIP DNA was quantified by quantitative PCR by the QuantiTect SYBR Green PCR Kit (Qiagen) using 7.5 μL of ChIP DNA and the ChIP PCR primers for mouse BRCA1 above. The PCR protocol consisted of 1 cycle of 900 seconds at 94°C followed by 40 cycles of (30 seconds at 94°C, 30 seconds at 60°C, and 30 seconds at 72°C).
Transient ChIP assay
MCF-10A cells were plated in serum-containing medium on 12-well culture dishes at a density of 5 × 104 cells per mL. After 24 hours, cells were transfected in triplicate with combinations of FLAG-GRα, GABPα, and GABPβ, and a series of BRCA1 promoter constructs. Cells were treated 24 hours after transfection with either ethanol vehicle or 1 μg/mL hydrocortisone in serum-free medium. ChIP was carried out as described earlier. Each reaction was carried out using chromatin from 2 × 106 cells and 1 μg per reaction of anti-FLAG antibody (sc-807; Santa Cruz Biotechnology Inc.) or no antibody as a negative control. Primers used during PCR analysis spanned the pRL vector insert [(+) 5′-GCAACGCGGCCTTTTTACGG and (−) 5′-CCTTAAACCTGTCTTGTAACC].
Results
GR upregulates BRCA1 promoter activity in the absence of ligand
We have previously shown that BRCA1 promoter activity is repressed in the presence of hydrocortisone in EPH-4 mouse mammary cells (19). To elucidate the mechanism through which this repression occurs, we evaluated the key stress signaling protein GR in regulating the expression of BRCA1 in breast cells. In transient cotransfection experiments in both EPH-4 mouse and 184-hTERT human mammary epithelial cells, overexpression of either the GRwt or a rat GR mutant that cannot bind DNA (GRL501P; ref. 29) resulted in a dramatic increase in human BRCA1 promoter (L6-pRL) activity in the absence of hydrocortisone (Fig. 1A, black bars). Treatment with hydrocortisone eliminated this increase in BRCA1 promoter activity (Fig. 1A, white bars). In the classically accepted model of GR function, hydrocortisone binds to GR in the cytoplasm, which subsequently translocates to the nucleus and regulates gene expression through specific elements or via bound transcription factors. In contrast, our observations suggest that GR acts as a positive regulator of BRCA1 in the nucleus in the absence of hydrocortisone ligand. To determine whether the endogenous GR behaves as an activator of BRCA1 expression, we transiently transfected EPH-4 cells with the BRCA1 promoter (L6-pRL) and an shRNA vector directed against mouse GR (shGR), which is able to reduce GR expression (Fig. 1B). Knockdown of GR by shGR repressed BRCA1 promoter activity in the absence but not in the presence of hydrocortisone (Fig. 1C), implicating GR as a positive regulator only in the absence of ligand.
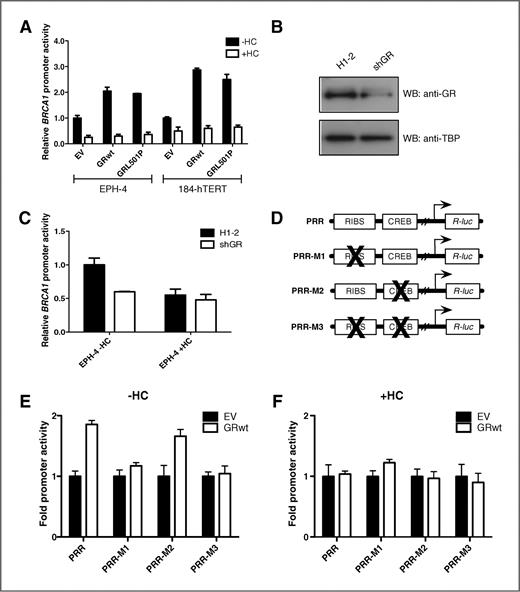
The unliganded GR is a positive regulator of BRCA1 expression. A, EPH-4 and 184-hTERT cells were transiently transfected with the L6 BRCA1 promoter reporter construct and expression vectors for wild-type rat GR (GRwt) and rat GR with a mutation of leucine to proline at amino acid position 501 (GRL501P), which abolishes its DNA-binding ability. Cells were treated 24 hours after transfection with either ethanol vehicle (−HC) or 1 μg/mL HC (+HC) and assayed for luciferase activity following a 48-hour incubation. B–C, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and either an empty vector (H1-2) or an shRNA vector directed against endogenous GR (shGR). Cells were (B) lysed after 48 hours and subjected to Western blotting (WB) to determine GR expression or (C) treated and assayed as above. D, schematic of BRCA1 promoter fragments (PRR, wild-type RIBS and CREB sites; PRR-M1, mutant RIBS; PRR-M2, mutant CREB; and PRR-M3, RIBS and CREB double mutant). E–F, EPH-4 cells were transiently transfected with a BRCA1 promoter fragment and an expression vector for wild-type rat GR (GRwt) in the (E) absence or the (F) presence of hydrocortisone (HC). Cells were treated and assayed as above. EV, empty vector.
The unliganded GR is a positive regulator of BRCA1 expression. A, EPH-4 and 184-hTERT cells were transiently transfected with the L6 BRCA1 promoter reporter construct and expression vectors for wild-type rat GR (GRwt) and rat GR with a mutation of leucine to proline at amino acid position 501 (GRL501P), which abolishes its DNA-binding ability. Cells were treated 24 hours after transfection with either ethanol vehicle (−HC) or 1 μg/mL HC (+HC) and assayed for luciferase activity following a 48-hour incubation. B–C, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and either an empty vector (H1-2) or an shRNA vector directed against endogenous GR (shGR). Cells were (B) lysed after 48 hours and subjected to Western blotting (WB) to determine GR expression or (C) treated and assayed as above. D, schematic of BRCA1 promoter fragments (PRR, wild-type RIBS and CREB sites; PRR-M1, mutant RIBS; PRR-M2, mutant CREB; and PRR-M3, RIBS and CREB double mutant). E–F, EPH-4 cells were transiently transfected with a BRCA1 promoter fragment and an expression vector for wild-type rat GR (GRwt) in the (E) absence or the (F) presence of hydrocortisone (HC). Cells were treated and assayed as above. EV, empty vector.
Promoter sites involved in GR-induced upregulation of BRCA1 promoter activity
We have previously shown that the RIBS, CREB (cAMP-responsive element binding protein), and UP (Upstream) promoter sites seem to play a role in the hydrocortisone-mediated regulation of BRCA1 expression, based on changes in complex formation in untreated and hydrocortisone-treated nuclear extracts (19). We concentrated on the RIBS element as it is a potent site of positive regulation and it contains a binding element for the heterodimeric ets transcription factor GABP. To characterize GR-responsive promoter elements, and to determine whether one or more of these sites are involved in the upregulation of BRCA1 by GR, EPH-4 cells were cotransfected with GRwt and various BRCA1 promoter mutant constructs (Fig. 1D). The addition of GRwt increased the activity of a construct containing only the RIBS and CREB sites (Fig. 1E, PRR) in the absence of ligand. Compared with the PRR promoter construct, mutation of the RIBS element (Fig. 1E, PRR-M1) eliminated activation whereas mutation of the CREB site (Fig. 1E, PRR-M2) did not. Mutation of both sites (Fig. 1E, PRR-M3) reduced BRCA1 upregulation to a level similar to that obtained with the PRR-M1 RIBS mutation construct alone. Therefore, RIBS is a key element in controlling BRCA1 expression in the presence of GR and the absence of ligand. In comparison, none of the BRCA1 mutant reporters were activated by GR in the presence of hydrocortisone ligand (Fig. 1F), suggesting that liganded GR is recruited away from the BRCA1 promoter.
GR interacts directly with the BRCA1 promoter
BRCA1 regulation by GR is not dependent on the DNA-binding ability of GR, as a GR mutant deficient in DNA binding (GRL501P) still activates BRCA1 expression in transient transfection assays (Fig. 1A, black bars). Furthermore, there are no defined GRE sequences within the BRCA1 promoter. Therefore, it is likely that GR exerts its effect on BRCA1 transcriptional activity by interacting with proteins present at the BRCA1 promoter, particularly at the RIBS element. ChIP analysis of endogenous GR in EPH-4 cells revealed that GR was present on the BRCA1 promoter in the absence of hydrocortisone (Fig. 2A, top). Transcription factors, GABPβ, Fra-2 (Fos-related AP1 transcription factor), and USF-2 (basic helix-loop-helix leucine zipper transcription factor), which are factors known to bind to the BRCA1 promoter through various elements, interacted with the BRCA1 promoter in the absence of hydrocortisone whereas the polycomb repressor RYBP did not (Fig. 2A, top). In the presence of hydrocortisone, GR dissociated from the promoter (Fig. 2A, bottom), as did GABPβ and Fra-2, which may be a result of loss of GR binding. USF-2 and RYBP bound BRCA1 in the presence of hydrocortisone (Fig. 2A, bottom). The ChIP DNA samples were analyzed via quantitative PCR and values obtained reflect the pattern of band intensities on the ChIP gel (Table 1). In general, there seems to be less transcription factor binding to BRCA1 in the presence of hydrocortisone. This and the fact that the repressor RYBP binds BRCA1 in this case may reflect the reduced activity of the BRCA1 promoter in the presence of hydrocortisone.

GR interacts directly with the BRCA1 promoter via the RIBS element only in the absence of hydrocortisone. A, chromatin was prepared from EPH-4 cells 24 hours after treatment with either ethanol vehicle (−HC) or 1 μg/mL hydrocortisone (+HC) in serum-free medium. ChIP analysis was carried out with the indicated antibodies and primers spanning the BRCA1 promoter. B, schematic of BRCA1 promoter constructs. C, MCF-10A cells were transiently transfected with each of the BRCA1 promoter constructs and different combinations of FLAG-tagged GR (FLAG-GRα), GABPα, and GABPβ. Cells were treated 24 hours after transfection with either ethanol vehicle or 1 μg/mL hydrocortisone in serum-free medium, and chromatin was prepared following a 24-hour incubation. The interaction of FLAG-GRα with elements of the BRCA1 promoter was examined through ChIP using FLAG antibody and subsequent PCR analysis of the immunoprecipitated DNA using primers spanning each promoter construct. Ab, antibody; GST, glutathione S-transferase; HA, hemagglutinin; H3, acetylated histone H3.
GR interacts directly with the BRCA1 promoter via the RIBS element only in the absence of hydrocortisone. A, chromatin was prepared from EPH-4 cells 24 hours after treatment with either ethanol vehicle (−HC) or 1 μg/mL hydrocortisone (+HC) in serum-free medium. ChIP analysis was carried out with the indicated antibodies and primers spanning the BRCA1 promoter. B, schematic of BRCA1 promoter constructs. C, MCF-10A cells were transiently transfected with each of the BRCA1 promoter constructs and different combinations of FLAG-tagged GR (FLAG-GRα), GABPα, and GABPβ. Cells were treated 24 hours after transfection with either ethanol vehicle or 1 μg/mL hydrocortisone in serum-free medium, and chromatin was prepared following a 24-hour incubation. The interaction of FLAG-GRα with elements of the BRCA1 promoter was examined through ChIP using FLAG antibody and subsequent PCR analysis of the immunoprecipitated DNA using primers spanning each promoter construct. Ab, antibody; GST, glutathione S-transferase; HA, hemagglutinin; H3, acetylated histone H3.
ChIP DNA products were analyzed by quantitative PCR and expressed as relative Ct values
| EPH-4 -HC | EPH-4 +HC | |
|---|---|---|
| Input | 34.41 | 35.58 |
| No Ab | >40 | >40 |
| GST | >40 | >40 |
| HA | >40 | >40 |
| H3 | 34.78 | 35.63 |
| GABPβ | 36.05 | >40 |
| Fra-2 | 35.15 | 37.4 |
| USF-2 | 34.52 | 36.02 |
| RYBP | >40 | 36.64 |
| GR | 35.7 | >40 |
| EPH-4 -HC | EPH-4 +HC | |
|---|---|---|
| Input | 34.41 | 35.58 |
| No Ab | >40 | >40 |
| GST | >40 | >40 |
| HA | >40 | >40 |
| H3 | 34.78 | 35.63 |
| GABPβ | 36.05 | >40 |
| Fra-2 | 35.15 | 37.4 |
| USF-2 | 34.52 | 36.02 |
| RYBP | >40 | 36.64 |
| GR | 35.7 | >40 |
To further characterize the promoter elements involved in the interaction between GR and the BRCA1 promoter, we conducted transient ChIP assays in nonmalignant MCF-10A human mammary epithelial cells. Cells were transiently transfected with a FLAG-tagged human GR construct, human GABPα and GABPβ, and various BRCA1 promoter mutant constructs (Fig. 2B). GR was immunoprecipitated with an anti-FLAG antibody to pull down GR, and BRCA1 constructs were detected via PCR using primers flanking the promoter inserts. FLAG-tagged GR coimmunoprecipitated the L6 BRCA1 construct only when exogenous GABPα and GABPβ were added (Fig. 2C, L6-pRL), indicating the requirement for this transcription factor in the interaction between GR and BRCA1, and showing the limiting quantities present in transient transfections. A RIBS multimeric construct (Fig. 2C, RIBSn-GF-TATA-pRL) and a BRCA1 promoter construct in which a number of important sites, but not the RIBS element, had been mutated (Fig. 2C, L6mUPmE2F-pRL) were also coimmunoprecipitated by GR in the presence of GABPα and GABPβ. In contrast, various RIBS deletion mutants were not immunoprecipitated by GR (Fig. 2C, L6ΔR-pRL, GF-TATA-pRL, and L6ΔRmUPmE2F-pRL). GR was found to interact with the RIBS promoter constructs only in the absence of hydrocortisone (Fig. 2C). When cells were treated with hydrocortisone, the interaction was abolished (Fig. 2C). In this context, these experiments show that the interaction between GR and the BRCA1 promoter is dependent on GABP and the RIBS element. Knockdown of GABP and subsequent transient ChIP would be ideal to establish the requirement for this transcription factor in the GR–BRCA1 interaction; however, knockdown of the GABP subunits results in significant cell death, limiting the ability to assess BRCA1 expression. Furthermore, BRCA1 expression is otherwise dependent on GABP binding outside of the GR context (11, 15). Because knockdown of GABP dramatically decreases BRCA1 expression, the assessment of GABP dependence in this context is not feasible. We have thus developed a novel model of BRCA1 regulation in which GR binds to the BRCA1 promoter via the RIBS site in the absence of hydrocortisone to activate expression, and in the presence of hydrocortisone, BRCA1 levels decrease due to the dissociation of GR from the promoter.
The interaction between GR and the BRCA1 promoter is mediated by GABPβ
Because GR interacts with the BRCA1 promoter through the GABP-binding RIBS element, and seems to require functional GABPα/β, its regulatory effect on BRCA1 may be via a protein–protein interaction with this multisubunit transcription factor. To determine whether GR interacts with GABP, we conducted coimmunoprecipitation experiments using lysates from human MCF-7 cells cotransfected with a FLAG-tagged GR construct along with untagged vectors for the α- and β-subunits of GABP. These experiments revealed that GABPβ, which contains the activation domain of the GABP protein, interacts with GR in the absence of hydrocortisone, both on its own and in combination with the DNA-binding α-subunit (Fig. 3A). Moreover, GR and GABPβ interact in the presence of hydrocortisone (Fig. 3B), suggesting that this interaction may be maintained during ligand binding to GR. This possibility is reflected by the ChIP results, which indicate that like GR, GABPβ interacts with BRCA1 only in the absence of hydrocortisone (Fig. 2A). In contrast, GR was not shown to interact with GABPα, either in the absence or presence of hydrocortisone.

GR interacts directly with the β-subunit of the ets transcription factor GABP. MCF-7 cells were transiently transfected with GABPα, GABPβ, and/or FLAG-GRα. Cells were treated 24 hours after transfection with either (A) ethanol vehicle (−HC) or (B) 1 μg/mL hydrocortisone (+HC) in serum-free medium, and whole-cell lysates were prepared following a 24-hour incubation. Lysates were immunoprecipitated (IP) with 1 μg of either anti-GABPβ or anti-FLAG antibody, and the presence of either FLAG (GR) or GABPβ in the eluted fractions was examined by Western blotting (WB). C and D, EPH-4 and MCF-7 cells were transiently transfected with GAL4 expression vectors for GABPα or GABPβ (GAL4-GABPα and GAL4-GABPβ, respectively) and expression vectors for both rat and human GR (GRwt and FLAG-GRα, respectively). Cells were treated 24 hours after transfection with either ethanol vehicle or 1 μg/mL hydrocortisone in serum-free medium and assayed for luciferase activity following a 24-hour incubation. E and F, EPH-4 and MCF-7 cells were transiently transfected with a GAL4 expression vector for GABPβ (pBIND-GABPβ FL) and a VP16 expression vector for human GR (pACT-GR FL). Cells were treated and assayed as above.
GR interacts directly with the β-subunit of the ets transcription factor GABP. MCF-7 cells were transiently transfected with GABPα, GABPβ, and/or FLAG-GRα. Cells were treated 24 hours after transfection with either (A) ethanol vehicle (−HC) or (B) 1 μg/mL hydrocortisone (+HC) in serum-free medium, and whole-cell lysates were prepared following a 24-hour incubation. Lysates were immunoprecipitated (IP) with 1 μg of either anti-GABPβ or anti-FLAG antibody, and the presence of either FLAG (GR) or GABPβ in the eluted fractions was examined by Western blotting (WB). C and D, EPH-4 and MCF-7 cells were transiently transfected with GAL4 expression vectors for GABPα or GABPβ (GAL4-GABPα and GAL4-GABPβ, respectively) and expression vectors for both rat and human GR (GRwt and FLAG-GRα, respectively). Cells were treated 24 hours after transfection with either ethanol vehicle or 1 μg/mL hydrocortisone in serum-free medium and assayed for luciferase activity following a 24-hour incubation. E and F, EPH-4 and MCF-7 cells were transiently transfected with a GAL4 expression vector for GABPβ (pBIND-GABPβ FL) and a VP16 expression vector for human GR (pACT-GR FL). Cells were treated and assayed as above.
We further investigated the interaction between GR and GABP through the use of a mammalian 2-hybrid system for which we created a mammalian expression vector consisting of the GAL4 DBD fused to GABPα or GABPβ, and used vectors for either GRwt or human GR (GRα) to complete the system. Cotransfection of the GAL4-GABPβ expression vector along with expression vectors for either GRwt or GRα in EPH-4 mouse (Fig. 3C) or MCF-7 human (Fig. 3D) mammary cells resulted in a dramatic increase in luciferase activity from a GAL4-responsive reporter in the absence of hydrocortisone, indicating an interaction between GABPβ and unliganded GR. The increase in activity of the GAL4-responsive promoter was presumed to be the result of the endogenous transactivation domain of GR, as the wild-type protein was used in place of a VP16 activation domain fusion. Treatment with hydrocortisone abolished the increase in luciferase activity, indicating that the transactivation by GR and GABPβ had been disrupted. No increase in luciferase activity was observed using GAL4-GABPα as a target (Fig. 3C and D), indicating that the α-subunit of GABP alone does not interact with GR. When a VP16 activation domain was attached to the GRα construct [pACT-GR full-length (FL)], a similar increase in reporter activity was observed in EPH-4 mouse (Fig. 3E) or MCF-7 human (Fig. 3F) mammary cells in only the presence of the GAL4-GABPβ fusion, and this activation was lost with hydrocortisone addition.
To further characterize the interaction between GR and GABPβ, we created a domain-specific deletion series of both GR and GABPβ based on the previously characterized domain structure of both proteins. The GR deletion mutants, including the full-length protein, and different combinations of the transcriptional activation domain (TAD), DBD, hinge region (HR), and ligand-binding domain (LBD), were cloned into a VP16 activation domain containing vector (pACT) to create pACT-GR fusions. Similarly, the GABPβ deletion mutants, including the full-length protein, the ankyrin repeat region (ANK), the region C-terminal to the ankyrin repeats (CT region), the activation domain (AD), and the leucine zipper region (LZ), were cloned into a GAL4 DBD containing vector (pBIND) to create pBIND-GABPβ fusions. Transient transfections involving each set of deletion mutants with the complementary full-length protein and a GAL4-responsive reporter revealed that the N-terminal to central region of GABPβ (including the activation domain) was involved in the interaction with the full-length GR (Fig. 4A–C, ANK-CT-AD). The increase in luciferase activity from the GAL4-responsive reporter is given as the ratio of each GABPβ deletion mutant plus full-length pACT-GR versus each mutant alone, to control for the basal activity of each fragment. Likewise, the N-terminal to central region (DBD and hinge region) of GR was involved in the interaction with GABPβ (Fig. 4D–F, TAD-DBD-HR). The increase in luciferase activity from the GAL4-responsive reporter is given as the ratio of each deletion mutant plus full-length pBIND-GABPβ versus each mutant alone.

The interaction between the GR and GABPβ requires the amino terminal to central regions of both proteins. EPH-4 and MCF-7 cells were transiently transfected with (A–C) full-length pACT-GR and various domain constructs of pBIND-GABPβ and (D–F) full-length pBIND-GABPβ and various domain constructs of pACT-GR. Cells were treated 24 hours after transfection with ethanol vehicle in serum-free medium and assayed for luciferase activity following a 24-hour incubation. GR domain abbreviations: TAD, transcriptional activation domain; DBD, DNA-binding domain; HR, hinge region; and LBD, ligand-binding domain. GABPβ domain abbreviations: ANK, ankyrin repeat region; CT, region C-terminal to the ankyrin repeats; AD, activation domain; and LZ, leucine zipper region.
The interaction between the GR and GABPβ requires the amino terminal to central regions of both proteins. EPH-4 and MCF-7 cells were transiently transfected with (A–C) full-length pACT-GR and various domain constructs of pBIND-GABPβ and (D–F) full-length pBIND-GABPβ and various domain constructs of pACT-GR. Cells were treated 24 hours after transfection with ethanol vehicle in serum-free medium and assayed for luciferase activity following a 24-hour incubation. GR domain abbreviations: TAD, transcriptional activation domain; DBD, DNA-binding domain; HR, hinge region; and LBD, ligand-binding domain. GABPβ domain abbreviations: ANK, ankyrin repeat region; CT, region C-terminal to the ankyrin repeats; AD, activation domain; and LZ, leucine zipper region.
To test whether GR lacking the entire ligand-binding region can activate the BRCA1 promoter, the pACT-GR fusion series, plus deletion mutants containing the DBD cloned into the pFLAG vector, were transiently transfected into EPH-4 cells along with the BRCA1 promoter. GR domain mutants, either VP16 (pACT) or FLAG (pFLAG) tagged, containing the N-terminal to central region of GR protein, specifically either the full-length (FL) or TAD-DBD-HR, activated BRCA1 in the absence of ligand (Fig. 5A). In a transient transfection of MCF-7 cells, both VP16 and FLAG-tagged versions of a GR mutant lacking the entire ligand-binding region (TAD-DBD-HR) activated BRCA1 in both the absence and presence of hydrocortisone, emphasizing that in contrast to the wild-type protein, it is now immune to the effects of hydrocortisone (Fig. 5B, GR TAD-DBD-HR black and white bars). This mutant is unable to bind ligand, but can still interact with GABPβ via its N-terminal to central hinge region, and activate BRCA1 expression via its N-terminal transactivation domain. This mutant also activated BRCA1 expression in the presence of titrated concentrations of the GR antagonist mifepristone (RU-486; Fig. 6A, GR TAD-DBD-HR) whereas the full-length GR did not (Fig. 6A, GR FL). RU-486 is a hydrocortisone analogue that is able to bind to GR but inhibits the transcription of GR target genes (31). As with hydrocortisone, treatment of mouse and human mammary cells with a comparable concentration of RU-486 [∼3 μmol/L hydrocortisone (1 μg/mL) compared with 5 μmol/L RU-486] resulted in decreased expression of endogenous BRCA1 (Fig. 6B), indicating that ligand binding, even of a nonsignaling ligand, to GR is the key physiologic stimulus for decreased BRCA1 expression. The repressive ability of RU-486 may be partly due to its capacity to disrupt unliganded GR binding at the promoters of its target genes.

GR domains activate the BRCA1 promoter in the absence of ligand binding. A, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and expression vectors for full-length FLAG-GR, full-length pACT-GR, and GR domain constructs either fused to VP16 (pACT) or without the VP16 fusion (pFLAG). Cells were treated 24 hours after transfection with ethanol vehicle in serum-free medium and assayed for luciferase activity following a 24-hour incubation. GR domain abbreviations are as in Fig. 4. B, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and both pFLAG and pACT expression vectors for each the full-length GR (GR FL) and GR lacking the LBD (GR TAD-DBD-HR) as well as empty vectors (pFLAG EV and pACT EV). Cells were treated 24 hours after transfection with either ethanol vehicle (−HC) or 1 μg/mL hydrocortisone (+HC) and assayed for luciferase activity following a 24-hour incubation.
GR domains activate the BRCA1 promoter in the absence of ligand binding. A, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and expression vectors for full-length FLAG-GR, full-length pACT-GR, and GR domain constructs either fused to VP16 (pACT) or without the VP16 fusion (pFLAG). Cells were treated 24 hours after transfection with ethanol vehicle in serum-free medium and assayed for luciferase activity following a 24-hour incubation. GR domain abbreviations are as in Fig. 4. B, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and both pFLAG and pACT expression vectors for each the full-length GR (GR FL) and GR lacking the LBD (GR TAD-DBD-HR) as well as empty vectors (pFLAG EV and pACT EV). Cells were treated 24 hours after transfection with either ethanol vehicle (−HC) or 1 μg/mL hydrocortisone (+HC) and assayed for luciferase activity following a 24-hour incubation.

GR antagonist RU-486 decreases BRCA1 expression. A, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and expression vectors for full-length GR (GR FL) and GR lacking the LBD (GR TAD-DBD-HR). Cells were treated 24 hours after transfection with either ethanol vehicle (UT) or 1 or 10 μmol/L RU-486 and assayed for luciferase activity following a 24-hour incubation. B, 184-hTERT, MCF-10A, and EPH-4 cells were treated 24 hours after plating with either ethanol vehicle (UT) or 5 μmol/L RU-486, and RNA was harvested following a 48-hour incubation. qRT-PCR analysis of endogenous BRCA1 expression was conducted using primers for either human or mouse BRCA1. BRCA1 Ct values were normalized to either HPRT1 (for 184-hTERT and MCF-10A) or TBP (EPH-4) internal control Ct values for triplicate samples and are presented as the level of expression in relation to untreated cells for each cell line.
GR antagonist RU-486 decreases BRCA1 expression. A, EPH-4 cells were transiently transfected with the L6 BRCA1 promoter reporter construct and expression vectors for full-length GR (GR FL) and GR lacking the LBD (GR TAD-DBD-HR). Cells were treated 24 hours after transfection with either ethanol vehicle (UT) or 1 or 10 μmol/L RU-486 and assayed for luciferase activity following a 24-hour incubation. B, 184-hTERT, MCF-10A, and EPH-4 cells were treated 24 hours after plating with either ethanol vehicle (UT) or 5 μmol/L RU-486, and RNA was harvested following a 48-hour incubation. qRT-PCR analysis of endogenous BRCA1 expression was conducted using primers for either human or mouse BRCA1. BRCA1 Ct values were normalized to either HPRT1 (for 184-hTERT and MCF-10A) or TBP (EPH-4) internal control Ct values for triplicate samples and are presented as the level of expression in relation to untreated cells for each cell line.
Discussion
BRCA1 has been associated with a wide variety of cellular functions, most recently the ability to regulate luminal breast cell differentiation and satellite DNA expression (9, 32). Regardless of the means by which BRCA1 blocks tumor formation, it is clear that its functional loss through mutational inactivation or by transcriptional downregulation leads to breast tumorigenesis. We have previously demonstrated that BRCA1 expression is negatively regulated by hydrocortisone (19) and suggested that this could provide a breast specific molecular mechanism to explain the increased risk associated with psychological stress (16). In this article, we have continued to explore the molecular mechanisms involved and have determined that, contrary to expectations, GR is actually a positive regulator of the BRCA1 promoter in the absence of ligand [Fig. 7 (1)]. This represents a novel mechanism of GR function and suggests that it plays a role in the basal transcription of the BRCA1 gene. Our original model suggested that long-term stress would result in the protracted elevation of cortisol levels and the consequent ongoing repression of BRCA1 [Fig. 7 (2)]. Although this is still a feasible model, these new results also imply that the basal levels of GR may play a role in determining BRCA1 levels. That is, reduced GR levels would lead to reduced transactivation of the BRCA1 promoter, which may be associated with an increased risk of breast cancer development. It has been reported that GR is differentially expressed in a variety of breast tissues, with strong expression in normal myoepithelial cells, and rare expression in nonmetaplastic carcinomas (33). Similarly, GR status has been negatively correlated with histologic tumor grade in estrogen receptor (ER)-negative/progesterone receptor (PR)-negative ductal intraepithelial neoplasia and invasive breast carcinoma tumors (34). Nuclear immunoreactivity to GR was found to decrease significantly with both tumor development and histologic grade (26, 34), suggesting a connection between functional GR levels and tumorigenesis. However, it has also been reported that GR expression is elevated in stage III infiltrating ductal carcinomas (35), and GR expression has also been correlated with patient age, with its expression being significantly higher in tumors from patients older than 50 years (36).
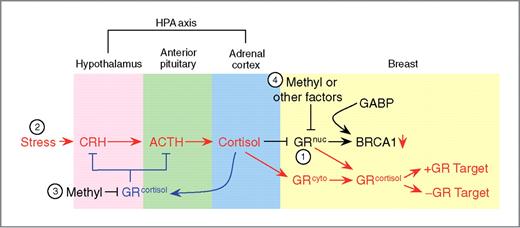
Model of GR regulation. 1, in the absence of stress (black), the unliganded nuclear GR (GRnuc) interacts with and positively regulates BRCA1 via GABP. 2, stress (red) increases production of corticotrophin-releasing hormone (CRH), which induces release of adrenocorticotropic hormone (ACTH), which results in cortisol secretion. The binding of cortisol to both nuclear and cytoplasmic GR (GRcyto) causes them (GRcortisol) to relocate to regular GR target genes. This results in reduced BRCA1 expression. The presence of cortisol negatively feeds back (blue) to both the hypothalamus and the pituitary, reducing the secretion of both CRH and ACTH. 3, as a result of prolonged stress exposure, methylation of the GR promoter in the brain decreases GR expression, resulting in loss of negative feedback at the hypothalamic-pituitary-adrenal (HPA) axis. Sustained production of cortisol results in loss of BRCA1-positive regulation by unliganded GR. 4, in breast tissue, systemic methylation of the GR promoter, or other factors, such as long-term glucocorticoid exposure and/or increased rate of protein turnover, leads to decreased GR expression, and subsequently to decreased BRCA1 activation.
Model of GR regulation. 1, in the absence of stress (black), the unliganded nuclear GR (GRnuc) interacts with and positively regulates BRCA1 via GABP. 2, stress (red) increases production of corticotrophin-releasing hormone (CRH), which induces release of adrenocorticotropic hormone (ACTH), which results in cortisol secretion. The binding of cortisol to both nuclear and cytoplasmic GR (GRcyto) causes them (GRcortisol) to relocate to regular GR target genes. This results in reduced BRCA1 expression. The presence of cortisol negatively feeds back (blue) to both the hypothalamus and the pituitary, reducing the secretion of both CRH and ACTH. 3, as a result of prolonged stress exposure, methylation of the GR promoter in the brain decreases GR expression, resulting in loss of negative feedback at the hypothalamic-pituitary-adrenal (HPA) axis. Sustained production of cortisol results in loss of BRCA1-positive regulation by unliganded GR. 4, in breast tissue, systemic methylation of the GR promoter, or other factors, such as long-term glucocorticoid exposure and/or increased rate of protein turnover, leads to decreased GR expression, and subsequently to decreased BRCA1 activation.
Our hypothesis that reduced GR levels may result in reduced BRCA1 expression is of particular interest given recent work suggesting that GR levels may be under long-term epigenetic regulation (37) and that repression of GR expression may be associated with specific stressful events occurring at various times during development (38). Both human and rat experiments have found that perinatal stress can result in methylation of the GR promoter in the brain [refs. 38, 39; Fig. 7 (3)], though it also appears that this may occur systemically, being detectable in peripheral blood (40). This raises the possibility that events in utero, perinatally, and/or during puberty in humans could result in the long-term reduction in GR levels either via systemic methylation of the GR promoter, or via any other mechanism which would bring about a decrease in GR levels, including long-term glucocorticoid exposure or increased rate of protein turnover [Fig. 7 (4)]. Reduction in GR levels would subsequently result in decreased BRCA1 levels due to the loss of positive regulation by GR (Fig. 7). These reduced BRCA1 levels would then be reflected in increased breast cancer risk. Elevated progesterone levels have been suggested to lead to an increase in the breast stem cell population (41, 42); altered BRCA1 levels as a consequence of altered GR levels may produce the same effect through decreased BRCA1 blocking differentiation.
We have shown that GR interacts with the β-subunit of the ets transcription factor GABP and that this interaction requires the N-terminal to central regions of both proteins (GR TAD-DBD-HR; GABPβ ANK-CT-AD). Several proteins have been reported to interact with GABPβ. The coactivator HCF binds the TAD of GABPβ via its central region to coordinate the assembly of the herpes simplex virus enhancer complex (43). The second zinc finger motif (CR2) of YEAF1 (YY1 and E4TF1/hGABP-associated factor 1) interacts with a small internal region of GABPβ to repress GABP-dependent transcription (44). The N-terminus of GABPβ interacts with the C-terminus of E2F1, including its transactivation and retinoblastoma protein (pRb)-binding domain (45). GR physically interacts with and negatively regulates the transcriptional activity of proteins such as NF-κB and activator protein (AP-1) via the second zinc finger of its DBD. In contrast, GR interacts with nuclear receptor coactivators of the p160 family as well as the CBP/p300 family mainly through its secondary activating function domain (AF2), located in the C-terminal LBD of the protein (46). Positive interactions with ligand-bound GR have been shown to occur through the LBD, and we have shown that the interaction between the unliganded GR and GABPβ is not dependent on the GR LBD. It is therefore possible that separate domains in GR may exist for targets of liganded and unliganded GR, whereas a region common to multiple protein interactions may be involved with GABPβ.
Classic models of GR signaling include ligand-induced binding to both positive and negative recognition elements (47) as well as directly binding and repressing other transcription factors such as NF-κB and AP-1 (48). The observation of a constitutive positive regulatory role for GR in the absence of ligand is unique and adds a new dimension to the GR pathway. A recent publication has reported negative regulation of the interleukin (IL)-6 gene by GR in the absence of ligand (25), but this effect is dependent on TNF-α and is thus not a constitutive change as we have observed. Indeed, reports of ligand-independent activation by other steroid hormone receptors have typically been in response to other stimuli (49). Our studies indicate that unliganded GR is recruited to promoters through its interaction with GABP. This suggests that a fourth class of GR-regulated genes exist, possibly overlapping with GABP target genes. ChIP-seq analysis of GR binding sites in A549 lung cells has revealed some 2,600 genes that are bound by unliganded GR, though with a much lower signal than liganded GR targets (50), suggesting this may be a widespread mechanism for gene regulation. The regulation of basal expression of target genes by unliganded GR suggests that in some contexts the endogenous levels of GR may be significant for gene regulation. That is, the level of unliganded nuclear GR in a given cell will help to determine the expression level of its target genes. This is in contrast to genes regulated by liganded GR where gene activation is dependent primarily on cortisol levels rather than GR levels. Given the wide degree of variability seen in GR protein levels across different cell types and its complex transcriptional regulation (with numerous 5′ exons; ref. 37), the regulation of genes by unliganded GR may be very particular to a particular tissue or cell.
Glucocorticoid signaling in the breast is associated with growth as well as the regulation of involution after lactation (51), where it seems to primarily regulate apoptosis (52). The nature of this regulation may actually change depending on the differentiation state, as dexamethasone induces cell-cycle inhibitors such as p21 in undifferentiated cells, whereas in differentiated cells it reduces their expression and inhibits apoptosis (53). The target genes of unliganded GR seem to be downregulated in the presence of glucocorticoids, suggesting that these genes may be opposing the function of liganded GR target genes. Given the known role of BRCA1 in regulating apoptosis (54), its induction by unliganded GR may represent a proapoptotic signal which is then lost in response to the antiapoptotic signal from hydrocortisone treatment. It is possible that the stress-induced downregulation of BRCA1, as well as other proapoptotic genes also regulated by unliganded GR, results in reduced monitoring and elimination of abnormal cells either during normal cellular turnover or during postlactational regression. This work provides new insight into the possible mechanisms responsible for the stress-induced increased risk of breast cancer as well as having wide implications for understanding the action of the glucocorticoids and GR.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
Conception and design: H.D. Ritter, L. Antonova, C.R. Mueller
Development of methodology: H.D. Ritter, L. Antonova, C.R. Mueller
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): H.D. Ritter, L. Antonova
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): H.D. Ritter, L. Antonova, C.R. Mueller
Writing, review, and/or revision of the manuscript: H.D. Ritter, L. Antonova, C.R. Mueller
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C.R. Mueller
Study supervision: C.R. Mueller
Acknowledgments
The authors thank Rachael Klinoski, Valerie Kelly-Turner, and Sherri Nicol for their excellent technical assistance, Dr. Calvin Roskelley for providing cell lines and the PRR constructs, as well as Dr. Keith Yamamoto for providing the GRwt and GRL501P constructs.
Grant Support
This work was funded by a grant from the Canadian Breast Cancer Foundation-Ontario Region, as well as a Doctoral Postgraduate Scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC PGS-D) to H.D. Ritter, a Student Fellowship from the Canadian Breast Cancer Foundation-Ontario Region to L. Antonova, and a Research Project Grant to C.R. Mueller from the Canadian Breast Cancer Foundation-Ontario Region.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.